This article was originally written in March of 1998 and appeared in RWJ Today during my first year of medical school. It is a good glimpse into my thinking at that time and the research background that I had come from at Cornell University:
The connection between blood cholesterol levels, dietary saturated fats, and vascular pathologies such as coronary artery disease and stroke is well known. Less well known, is the link between blood homocysteine levels and these same vascular diseases.
Homocysteine is an amino acid that is normally remethylated by methionine synthase, a vitamin B12 and folate-dependent enzyme, to produce methionine. Methionine is then utilized for protein synthesis directly, and indirectly as S-adenosylmethionine, for a multitude of methylation reactions throughout the cell. Disruption in methionine synthase activity, either by genetic mutation or cofactor deficiency, causes abnormal buildup of plasma homocysteine levels.
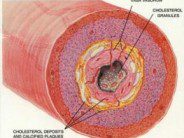
An alternative route for homocysteine disposal is via the vitamin B6 dependent enzyme, cystathionine beta synthase. This enzyme uses serine and homocysteine to produce cystathionine. Cystathionine is then converted to cysteine in the liver. Disruptions in this enzymatic activity also cause elevated plasma homocysteine levels. Elevated plasma homocysteine levels can then cause damage to intimal cells of the blood vessels by modification of low-density lipoprotein (LDL) by homocysteine thiolactone, which causes LDL self-aggregation and subsequent phagocytosis by macrophages. These macrophages form foam cells, which lead to deposition of cholesterol and lipids, oxidative damage, thrombogenesis, and development of arteriosclerotic plaques.
The first hint of homocysteine’s role in arteriosclerosis came in 1933 when an eight-year old boy presented with headache, drowsiness, and vomiting at the Massachusetts General Hospital. In addition to these findings, it was found that his urine contained abnormally high levels of homocysteine. After admission, the boy showed signs of elevated temperature and blood pressure without infection. His condition deteriorated rapidly and signs of a right-sided stroke appeared. After three days, he died. It was later shown that he had a condition called homocysteinuria, caused by homozygous mutations of his genes encoding cystathionine beta synthase, which was the cause of his elevated urine homocysteine levels. The pathologist found the cause of death to be arteriosclerosis of the carotid artery with cerebral infarct.
A similar but rarer for of homocysteinuria in a two-month old boy, which presented some years later also hinted to homocysteine’s role in arteriosclerosis. At the time of death, this two-month old boy was found to have elevated plasma and urine homocysteine levels and advanced arteriosclerosis. It was later determined that this case of homocysteinuria was caused by homozygous mutation of his genes encoding methionine synthase.
Thus, two separate congenital and genetic defects in homocysteine metabolism both conferred increased susceptibility to the development of arteriosclerosis. By any scientific standard, these two observations would not be enough to conclusively link homocysteine to arteriosclerosis, but they did establish the groundwork for further investigation. In fact, since the publication of those initial observations, accumulating data obtained by different lines of experimentation have consistently confirmed homocysteine’s role in the pathogenesis of arteriosclerosis. The most recent study, published in the February 4 issue of the Journal of the American Medical Association (JAMA), found that dietary intake of folate and vitamin B6 reduced mortality and morbidity form cardiovascular disease during a 14 –year period among women.
As the general population does not suffer from homozygous or heterozygous mutations of either methionine synthase or cystathionine beta synthase, one could argue that these findings, although relevant to genetic hyperhomocysteinuria, do not relate to arterioslcerosis as found within the normal progression of the disease. Yet, a third to a half of all cases of arteriosclerosis have normal cholesterol levels. Thus, other factors must play a significant role in the development and etiology of the disease. These other factors include elevated plasma homocysteine caused by environmental factors, such as vitamin deficiency, instead of genetic enzymatic mutation.
The Food and Drug Administration has recently approved the fortification of the American food supply with folate to prevent the occurrence of neural tube defects such as anencephaly and spina bifida. This strategy may also prevent or delay the progression of arteriosclerosis because a deficiency of folate, vitamin B6, and vitamin B12 is implicated in the development of hyperhomocysteinuria.
References:
- McCully, KS (1969) “Vascular pathology of homocysteinuria: Implications for the pathogenesis of arteriosclerosis.” Am. J. Pathol. 56, 111-128.
- McCully, KS (1997) The Homocysteine Revolution: Medicine for the New Millennium. New Canaan, Conn.: Keats Publishing.
- Rimm, EB, Willet, WC, Hu, FB, et. al. (1998) “Folate and vitamin B6 from diet and supplements in relation to risk of coronary heart disease among women.” JAMA 279, 359-364.







 DrSamGirgis.com is a blog about medicine, nutrition, health, wellness, and breaking medical news. At DrSamGirgis.com, the goal is to provide a forum for discussion on health and wellness topics and to provide the latest medical research findings and breaking medical news commentary.
DrSamGirgis.com is a blog about medicine, nutrition, health, wellness, and breaking medical news. At DrSamGirgis.com, the goal is to provide a forum for discussion on health and wellness topics and to provide the latest medical research findings and breaking medical news commentary.
{ 11 comments… read them below or add one }
If you could e-mail me with a few suggestions on just how you made your blog look this excellent, I would be grateful.
Many thanks for writing
Hello, good article, I just set your website in my favourites so I can keep following your page. Thanks.
You have a few useful suggestions on this web site.
very good information you write it very clean. I’m very lucky to get this info from you.
An impressive share, I just given this onto a colleague who was doing a little analysis on this. And he in fact bought me breakfast because I found it for him.. smile. So let me reword that: Thnx for the treat! But yeah Thnkx for spending the time to discuss this, I feel strongly about it and love reading more on this topic. If possible, as you become expertise, would you mind updating your blog with more details? It is highly helpful for me. Big thumb up for this blog post!
I must admit this post is very wonderful . Thanks once again for the push!
It’s exhausting to seek out knowledgeable individuals on this matter, but you sound like you understand what you’re talking about! Thanks
Good day! I simply wish to give an enormous thumbs up for the nice data you’ve gotten here on this post. I can be coming back to your blog for extra soon.
Continue to keep the best level and tell us more useful information.Thanks.
L-Cysteine is an amino acid that is closely related to Cystine. Cystine contains sulfur and is formed by two molecules of L-Cysteine. L-Cysteine is also a sulfur containing amino acid. It is used to manufacture L-glutathione and L-taurine.’*;’:
Kind regards
{ 1 trackback }